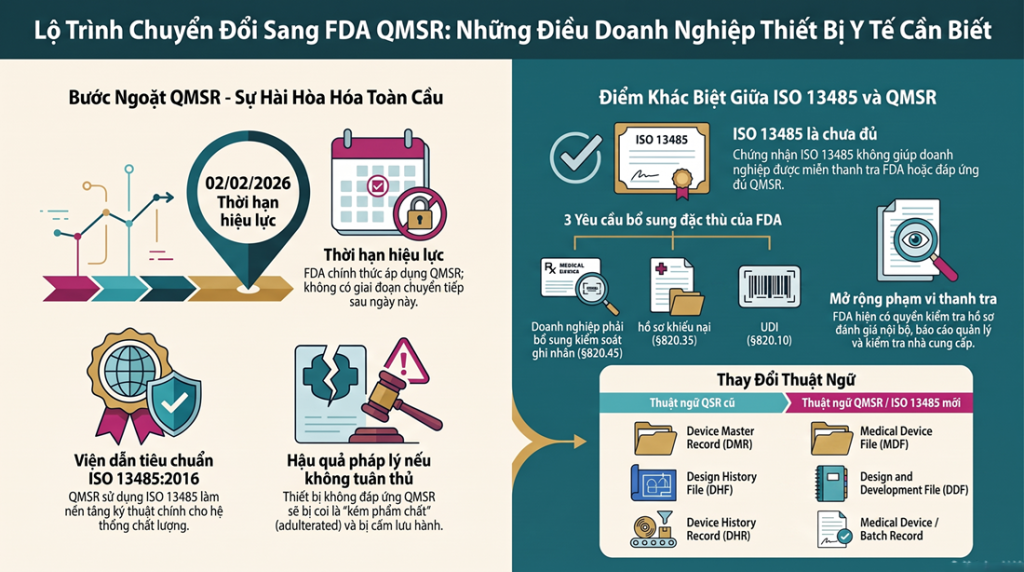
Sau khi 21 CFR Part 820 chuyển thành QMSR và viện dẫn ISO 13485:2016 (hiệu lực 02/02/2026), nhiều doanh nghiệp lầm tưởng “có ISO 13485 là xong”. Thực tế, FDA giữ lại và bổ sung một số yêu cầu riêng mà ISO 13485 không bao phủ đầy đủ. Bài viết này phân tích chi tiết các yêu cầu cốt lõi của QMSR, so sánh QSR cũ với QMSR và chỉ rõ những điểm FDA bắt buộc thêm.
Bức tranh tổng thể: QMSR = ISO 13485:2016 + Yêu cầu riêng của FDA
QMSR lấy ISO 13485:2016 làm nền tảng cho toàn bộ hệ thống quản lý chất lượng (QMS), nhưng chồng lên đó là các quy định đặc thù Hoa Kỳ. Nói cách khác:
Tuân thủ QMSR = Đáp ứng ISO 13485:2016 + Đáp ứng các điều khoản bổ sung tại Part 820 (§820.10, §820.35, §820.45 …) + Liên kết tới các quy định FDA khác (Part 803, 806, 821, 830).
Các yêu cầu nền tảng theo ISO 13485:2016
Vì QMSR viện dẫn ISO 13485:2016, doanh nghiệp phải đáp ứng đầy đủ các nhóm yêu cầu của tiêu chuẩn này, bao gồm:
- Hệ thống quản lý chất lượng và cách tiếp cận dựa trên rủi ro (risk-based approach).
- Trách nhiệm lãnh đạo và xem xét của lãnh đạo (management review).
- Quản lý nguồn lực, năng lực và đào tạo nhân sự, môi trường làm việc.
- Hoạch định và kiểm soát thiết kế & phát triển (Design & Development – Điều 7).
- Kiểm soát mua hàng và nhà cung cấp.
- Sản xuất và cung cấp dịch vụ, xác nhận giá trị sử dụng quy trình (process validation), truy xuất nguồn gốc.
- Đo lường, phân tích, cải tiến: đánh giá nội bộ, kiểm soát sản phẩm không phù hợp, hành động khắc phục – phòng ngừa (CAPA).
Một thay đổi đáng chú ý: từ “rủi ro (risk)” gần như không xuất hiện trong QSR cũ, nhưng được nhấn mạnh xuyên suốt trong ISO 13485:2016. QMSR vì thế codified (luật hóa) kỳ vọng quản lý rủi ro trong toàn bộ QMS.
Những điểm FDA bổ sung ngoài ISO 13485 (quan trọng nhất)
1. §820.10 – Yêu cầu đối với QMS và “applicable regulatory requirements”
Đây là mục then chốt. §820.10(b) liên kết các yêu cầu FDA bắt buộc bổ sung để tuân thủ đầy đủ các điều khoản ISO 13485, gồm:
- Báo cáo thiết bị y tế (MDR) – 21 CFR Part 803.
- Khắc phục và thu hồi (Corrections and Removals) – 21 CFR Part 806.
- Theo dõi thiết bị (Device Tracking) – 21 CFR Part 821.
- Nhận dạng thiết bị duy nhất (UDI) – 21 CFR Part 830.
§820.10(c) làm rõ rằng thiết bị theo diện IDE không được miễn yêu cầu kiểm soát thiết kế (Design & Development – Điều 7 ISO 13485). §820.10(d) yêu cầu truy xuất thiết bị cấy ghép/duy trì sự sống theo Điều 7.5.9.2 ISO 13485 — chặt hơn QSR cũ (vốn chỉ tới người nhận đầu tiên).
2. §820.35 – Kiểm soát hồ sơ (Control of Records)
Bổ sung yêu cầu nội dung chi tiết hơn ISO 13485 đối với:
- Hồ sơ khiếu nại: khi nào phải điều tra, thông tin bắt buộc (mã UDI/UPC, thông tin người khiếu nại…), liên kết nghĩa vụ báo cáo theo Part 803.
- Hồ sơ dịch vụ (servicing records).
- Tài liệu đáp ứng UDI và bảo mật hồ sơ gửi/nhận FDA.
3. §820.45 – Kiểm soát ghi nhãn và bao bì thiết bị
FDA cho rằng ISO 13485 chưa đủ về kiểm soát ghi nhãn/bao bì, nên bổ sung yêu cầu kiểm tra độ chính xác của nhãn trước khi xuất xưởng (tương tự §820.120(b) trong QSR cũ).
So sánh nhanh QSR (cũ) và QMSR (mới)
| Tiêu chí | QSR (trước 02/02/2026) | QMSR (từ 02/02/2026) |
|---|---|---|
| Tên gọi | Quality System Regulation | Quality Management System Regulation |
| Cơ sở | Văn bản FDA tự viết | Viện dẫn ISO 13485:2016 + định nghĩa ISO 9000 |
| Độ dài | Dài, chi tiết từng yêu cầu | Ngắn hơn, nhiều mục “Reserved” |
| Quản lý rủi ro | Xuất hiện rất hạn chế | Xuyên suốt toàn QMS |
| Thanh tra | Kỹ thuật QSIT | Compliance Program 7382.850 (bỏ QSIT) |
| Hài hòa quốc tế | Khung riêng của Mỹ | Đồng bộ ISO/MDSAP |
Thanh tra và MDSAP – những lưu ý thực tế
- FDA vẫn giữ thẩm quyền thanh tra sau 02/02/2026; chỉ thay đổi quy trình (bỏ QSIT, dùng 7382.850).
- Chứng nhận ISO 13485 không miễn trừ thanh tra FDA.
- Doanh nghiệp tham gia MDSAP tiếp tục được miễn thanh tra định kỳ thường quy của FDA.
- QMSR cho phép FDA xem xét hồ sơ xem xét lãnh đạo, đánh giá chất lượng và báo cáo đánh giá nhà cung cấp (các ngoại lệ trước đây tại §820.180(c) không còn được duy trì).
Lưu ý cho sản phẩm phối hợp (combination products)
QMSR có các chỉnh sửa đồng bộ tại 21 CFR Part 4 để làm rõ yêu cầu QMS thiết bị y tế của sản phẩm phối hợp, nhưng không thay đổi yêu cầu CGMP đối với sản phẩm phối hợp.
Câu hỏi thường gặp (FAQ)
1. Doanh nghiệp đã đạt ISO 13485:2016 cần bổ sung gì cho QMSR? Chủ yếu bổ sung các yêu cầu FDA tại §820.10 (MDR, UDI, theo dõi, khắc phục/thu hồi), §820.35 (hồ sơ khiếu nại/dịch vụ) và §820.45 (kiểm soát nhãn/bao bì).
2. QMSR có yêu cầu người xem xét thiết kế độc lập không? ISO 13485 không bắt buộc người xem xét độc lập trong thiết kế & phát triển; tuy nhiên doanh nghiệp cần tham chiếu kỳ vọng của FDA nêu trong preamble của quy định.
3. Hồ sơ tạo trước 02/02/2026 có còn giá trị không? Có. FDA có thể xem xét hồ sơ QMS tạo trước ngày hiệu lực. Doanh nghiệp nên thực hiện phân tích đối chiếu để chứng minh hồ sơ cũ đáp ứng yêu cầu QMSR.
4. QSIT bị bỏ thì FDA thanh tra thế nào? FDA dùng quy trình trong Compliance Program 7382.850; bản chất vẫn là thanh tra hệ thống chất lượng tương tự trước đây.
Liên hệ tư vấn đáp ứng QMSR & ISO 13485:2016
Chúng tôi giúp doanh nghiệp rà soát khoảng cách QSR → QMSR, bổ sung đúng các yêu cầu FDA và sẵn sàng cho thanh tra.
STC VN Co., Ltd. (Staunchly Vietnam)
📞 Hotline: +84 933 096 426 – +84 868 591 260
✉️ Email: info@staunchlyservices.com.vn
🌐 Website: staunchlyservices.com.vn

LỘ TRÌNH CHIẾN LƯỢC: CHUYỂN ĐỔI TỪ QSR SANG QMSR (21 CFR PART 820)
1. Bối cảnh Chiến lược và Sự trỗi dậy của QMSR
Việc FDA ban hành quy định cuối cùng về Quản lý Hệ thống Chất lượng (QMSR) không chỉ là một đợt cập nhật văn bản thông thường, mà là cuộc cải cách sâu rộng nhất trong hơn 25 năm qua đối với ngành thiết bị y tế toàn cầu. Bằng cách viện dẫn trực tiếp tiêu chuẩn ISO 13485:2016 vào 21 CFR Part 820, FDA đã chính thức tạo ra một “ngôn ngữ chung” cho quản lý chất lượng. Điều này cho phép các doanh nghiệp giảm bớt gánh nặng duy trì các hệ thống song song, tiết kiệm ước tính 532 triệu USD cho toàn ngành và thúc đẩy sự hội nhập hoàn toàn vào chương trình thanh tra MDSAP.
Tuy nhiên, với tư cách là chuyên gia tư vấn, tôi cần nhấn mạnh một thông điệp khẩn cấp: Sẽ không có giai đoạn ân hạn (no grace period). Đúng ngày 02/02/2026, mọi hệ thống chất lượng của doanh nghiệp phải vận hành theo QMSR. Bất kỳ sự chậm trễ nào cũng sẽ khiến thiết bị bị coi là “kém phẩm chất” (adulterated) theo Mục 501(h) của Đạo luật FD&C, dẫn đến rủi ro bị đình chỉ lưu hành hoặc từ chối nhập khẩu ngay lập tức tại cửa khẩu Hoa Kỳ. Việc nắm vững sự thay đổi thuật ngữ và cấu trúc là bước đi chiến lược đầu tiên để tồn tại trong kỷ nguyên mới này.
2. Phân tích Sự thay đổi Hệ thống: Từ Thuật ngữ đến Cấu trúc
Sự dịch chuyển sang QMSR yêu cầu một đợt “tổng rà soát” và tái cấu trúc kiến trúc tài liệu. Doanh nghiệp cần đặc biệt lưu ý rằng một số thuật ngữ cũ đã bị loại bỏ hoàn toàn về mặt pháp lý, mặc dù các yêu cầu về dữ liệu vẫn được duy trì dưới những tên gọi mới.
| Thuật ngữ cũ (QSR) | Thuật ngữ mới (QMSR/ISO 13485) | Phân tích tác động chiến lược |
| DHF (Design History File) | DDF (Design and Development File) | Duy trì hồ sơ chứng minh thiết kế theo Điều 7.3 của ISO 13485. |
| DMR (Device Master Record) | MDF (Medical Device File) | MDF có phạm vi rộng hơn DMR. Ngoài thông số sản xuất, MDF phải bao gồm mục đích sử dụng (intended use) và ghi nhãn (theo ISO 4.2.3). |
| DHR (Device History Record) | Medical Device/Batch Record | Thuật ngữ “DHR” đã chết về mặt pháp lý, nhưng hồ sơ lô vẫn là bắt buộc để chứng minh tính truy xuất nguồn gốc. |
| Management Representative | Top Management | Dịch chuyển trách nhiệm từ cá nhân (Đại diện lãnh đạo) sang Ban lãnh đạo cấp cao nhất (CEO/HĐQT). |
Phân tích lớp “So What?” – Tại sao Ban lãnh đạo phải hành động? Thay đổi quan trọng nhất nằm ở khái niệm “Top Management”. FDA không còn chấp nhận việc ủy quyền hoàn toàn trách nhiệm chất lượng cho một cá nhân đơn lẻ. CEO hiện nay là đối tượng trực tiếp chịu trách nhiệm về chính sách chất lượng và nguồn lực. Trong các đợt thanh tra mới, FDA sẽ tập trung vào sự tham gia thực tế của lãnh đạo trong các buổi họp xem xét hệ thống. Một Ban lãnh đạo thờ ơ sẽ là mục tiêu hàng đầu cho các quan sát lỗi (483) nghiêm trọng nhất.
Lưu ý đặc biệt về định nghĩa Rework (§820.3(b)): Doanh nghiệp cần phân biệt rõ, FDA đã bác bỏ định nghĩa “Rework” của ISO 9000. Theo QMSR, Rework chỉ được tính là hành động khắc phục thực hiện trước khi sản phẩm được xuất xưởng. Mọi hoạt động sau khi phân phối sẽ được coi là hiệu chỉnh hoặc thu hồi (recall) dưới các quy định khác.
3. “Lớp bổ sung của FDA”: Những yêu cầu nằm ngoài ISO 13485:2016
Một sai lầm chí tử mà nhiều doanh nghiệp Việt Nam và FDI thường mắc phải là lầm tưởng rằng “có chứng chỉ ISO 13485 là mặc nhiên tuân thủ QMSR”. FDA duy trì một bộ các yêu cầu “anchors” (neo giữ) để bảo vệ tính thực thi của Đạo luật FD&C mà ISO không có.
03 khu vực bổ sung bắt buộc của FDA:
- Kiểm soát hồ sơ (§820.35): Đây là nơi FDA yêu cầu chi tiết hơn ISO.
- Hồ sơ khiếu nại (Complaints): Phải chứa 07 trường thông tin, trong đó có các trường khó như: mã UDI/UPC, số điện thoại của người khiếu nại, và đặc biệt là văn bản phản hồi cho người khiếu nại.
- Hồ sơ dịch vụ (Service): Phải chứa 06 trường thông tin cụ thể về dữ liệu kiểm tra và người thực hiện.
- Ngôn ngữ: Mặc dù không ghi trong QMSR, nhưng FDA yêu cầu mọi hồ sơ thanh tra phải ở dạng mà điều tra viên có thể sử dụng (thường là tiếng Anh hoặc có bản dịch sẵn sàng).
- Ghi nhãn và Bao bì (§820.45): FDA đòi hỏi một quy trình kiểm soát khắt khe hơn: phải thực hiện kiểm tra độ chính xác của nhãn so với MDF trước khi xuất xưởng để ngăn ngừa lỗi dán nhãn sai – nguyên nhân hàng đầu gây thu hồi sản phẩm.
- Liên kết hệ thống (§820.10): Hệ thống QMS của bạn phải “neo” chặt với các quy định khác:
- Hệ thống Định danh thiết bị duy nhất (UDI – Part 830).
- Báo cáo thiết bị y tế (MDR – Part 803).
- Theo dõi thiết bị (Tracking – Part 821) cho các thiết bị cấy ghép hoặc hỗ trợ sự sống.
4. Cải cách Thanh tra: Sự kết thúc của quyền miễn trừ hồ sơ
Sự thay đổi quan trọng nhất từ tháng 2/2026 không nằm ở tài liệu, mà ở cách FDA thanh tra bạn. Kỹ thuật thanh tra QSIT cũ sẽ bị thay thế bởi chương trình Compliance Program 7382.850, bám sát cấu trúc của ISO 13485.
Điểm thay đổi cốt lõi về quyền tiếp cận dữ liệu: Dưới thời QSR, doanh nghiệp được hưởng quyền miễn trừ theo §820.180(c) – nghĩa là FDA không được xem xét các báo cáo đánh giá nội bộ (internal audits) và báo cáo xem xét của lãnh đạo (management reviews). Quyền này đã bị bãi bỏ trong QMSR.
Đánh giá tác động: Hiện nay, FDA có quyền tiếp cận đầy đủ các báo cáo đánh giá nội bộ, báo cáo xem xét của lãnh đạo và cả báo cáo đánh giá nhà cung cấp (supplier audits). Điều này đòi hỏi sự minh bạch tuyệt đối. Ban lãnh đạo không thể che giấu các lỗ hổng hệ thống. Nếu một lỗi nghiêm trọng được phát hiện trong đánh giá nội bộ nhưng doanh nghiệp không có hành động khắc phục (CAPA) thỏa đáng, đó sẽ là bằng chứng không thể chối cãi để FDA ban hành Warning Letter.
5. Lộ trình 8 Bước: Chuyển đổi và Tuân thủ QMSR
Để sẵn sàng cho mốc thời gian 02/02/2026, doanh nghiệp cần một kế hoạch hành động quyết liệt:
Bước 1: Phân tích khoảng cách (Gap Analysis)
Thực hiện ma trận đối chiếu (mapping) giữa hệ thống QSR hiện tại, ISO 13485 và các phần bổ sung tại §820.10, §820.35, §820.45.
Bước 2: Hoạch định hệ thống và MDF
Thiết kế lại kiến trúc tài liệu. Xây dựng Medical Device File (MDF) thay thế cho DMR, đảm bảo tích hợp đầy đủ mục đích sử dụng và các tham chiếu rủi ro.
Bước 3: Cập nhật quy trình vận hành (SOPs)
Tích hợp các yêu cầu cụ thể về trường thông tin hồ sơ khiếu nại/dịch vụ và quy trình kiểm soát nhãn trước xuất xưởng vào hệ thống hiện hành.
Bước 4: Quản lý rủi ro thực chất (ISO 14971)
FDA kỳ vọng rủi ro không chỉ nằm ở hồ sơ thiết kế. Doanh nghiệp phải áp dụng tư duy rủi ro xuyên suốt từ kiểm soát quá trình, lựa chọn nhà cung cấp đến phản hồi sau thị trường theo framework của ISO 14971.
Bước 5: Đào tạo và Văn hóa Chất lượng
Nâng cao nhận thức cho mọi cấp độ. Đặc biệt là đào tạo cho Ban lãnh đạo về trách nhiệm pháp lý mới khi hồ sơ xem xét lãnh đạo bị FDA soi xét.
Bước 6: Xác nhận giá trị sử dụng (Validation)
Thực hiện xác nhận lại các quy trình sản xuất và phần mềm quản lý chất lượng (eQMS) nếu có thay đổi trong cấu trúc vận hành mới.
Bước 7: Đánh giá thử nghiệm (Mock Inspection)
Thực hiện đánh giá nội bộ theo phương pháp của Compliance Program 7382.850. Đây là lúc kiểm tra khả năng giải trình các hồ sơ trước đây vốn được “bảo mật” trước FDA.
Bước 8: Chốt chặn tuân thủ và Sẵn sàng thanh tra
Hoàn thiện hồ sơ sẵn sàng cho các đợt thanh tra không báo trước từ FDA hoặc các tổ chức MDSAP.
6. Quản lý Rủi ro và Văn hóa Chất lượng: Trọng tâm của QMSR
Triết lý của FDA trong QMSR là: “Chất lượng phải được xây dựng vào sản phẩm, không phải được kiểm tra vào sản phẩm” (Quality must be built into the product, not inspected in). Sự dịch chuyển sang tư duy rủi ro chủ động là bắt buộc.
FDA sẽ đánh giá “Văn hóa Chất lượng” của doanh nghiệp thông qua các bằng chứng hữu hình:
- Sự nhạy bén của CAPA: Doanh nghiệp có chủ động tìm lỗi tiềm ẩn (Preventive Action) hay chỉ đợi lỗi xảy ra mới sửa (Corrective Action)?
- Trách nhiệm nhà cung cấp: Bạn kiểm soát rủi ro từ linh kiện mua ngoài như thế nào?
- Phản hồi sau thị trường (§8.2.1): Doanh nghiệp có hệ thống thu thập thông tin “feedback” chủ động hay chỉ ngồi chờ khiếu nại?
Một “văn hóa chất lượng yếu” thường bộc lộ qua một chuỗi các quan sát 483 lặp lại trong các kỳ xem xét của lãnh đạo và sự chậm trễ trong việc đóng các CAPA quan trọng.
7. Khuyến nghị và Kết luận: Tương lai của sự hài hòa hóa
QMSR chính là tấm vé thông hành để doanh nghiệp MedTech bước vào sân chơi toàn cầu với một tiêu chuẩn duy nhất. Để không bị đào thải khỏi thị trường Hoa Kỳ sau tháng 2/2026, tôi đưa ra 03 khuyến nghị chiến lược:
- Hành động ngay lập tức: Thời gian 2 năm chuẩn bị đang trôi đi nhanh chóng. Việc trì hoãn sẽ dẫn đến tình trạng “adulterated” và import refusal – những án phạt có thể phá hủy uy tín doanh nghiệp.
- Tập trung vào hồ sơ thiết kế (DDF): Đây là nền tảng cốt yếu nhất chứng minh an toàn và hiệu quả theo tiêu chuẩn mới.
- Xây dựng hệ thống QMS điện tử (eQMS): Việc quản lý hồ sơ MDF và DDF phức tạp trên giấy sẽ trở thành điểm yếu lớn khi FDA thanh tra theo phương thức mới. Một hệ thống điện tử giúp truy xuất dữ liệu trong vài giây thay vì vài giờ là lợi thế cạnh tranh sống còn.
Tuân thủ QMSR không chỉ là để vượt qua một kỳ kiểm tra pháp lý, mà là lời cam kết cao nhất của doanh nghiệp trong việc bảo vệ sức khỏe người bệnh thông qua những sản phẩm đạt chuẩn quốc tế tối ưu.
Hướng dẫn Kỹ thuật: Thiết lập và Duy trì Hồ sơ Thiết bị Y tế theo Tiêu chuẩn QMSR và ISO 13485:2016
1. Tổng quan về Sự chuyển đổi sang QMSR (Quality Management System Regulation)
Kể từ ngày 02/02/2026, FDA chính thức áp dụng quy định QMSR, thay thế toàn diện quy định QSR cũ. Đây là bước ngoặt lớn nhất trong hơn 25 năm qua, chuyển dịch từ một tiêu chuẩn riêng biệt của Hoa Kỳ sang việc viện dẫn bằng tham chiếu (IBR) tiêu chuẩn quốc tế ISO 13485:2016.
Cảnh báo chiến lược: FDA đã khẳng định không có giai đoạn chuyển tiếp (No Grace Period) sau ngày 02/02/2026. Bất kỳ thiết bị y tế nào không tuân thủ QMSR sau cột mốc này sẽ bị coi là “adulterated” (kém phẩm chất) theo Mục 501(h) của Đạo luật FD&C. Điều này có thể dẫn đến các biện pháp chế tài nghiêm khắc như thư cảnh báo (Warning Letter), thu hồi sản phẩm, hoặc cấm nhập khẩu vào thị trường Hoa Kỳ.
Nguyên tắc ưu tiên định nghĩa: Trong kỷ nguyên QMSR, các chuyên gia RA/QA phải nắm vững thứ tự ưu tiên pháp lý khi có xung đột thuật ngữ:
- Đạo luật FD&C: Ưu tiên tối cao (ví dụ: định nghĩa “Device”, “Labeling”).
- QMSR (§820.3): Các định nghĩa FDA giữ lại để bổ sung cho ISO (ví dụ: “Component”, “Finished Device”).
- ISO 13485:2016.
- ISO 9000:2015.
Lưu ý đặc biệt về “Rework” (Gia công lại): Doanh nghiệp cần đặc biệt chú ý định nghĩa Rework tại §820.3. FDA từ chối sử dụng định nghĩa của ISO 9000 vì QMSR giới hạn Rework chỉ là các hành động khắc phục thực hiện trước khi phân phối. Các hành động sau phân phối sẽ được quản lý theo các điều khoản về thu hồi hoặc báo cáo thiết bị y tế.
2. Bản đồ Chuyển đổi Thuật ngữ và Cấu trúc Hồ sơ mới
Việc chuyển đổi thuật ngữ không đơn thuần là thay đổi danh xưng, mà là sự thay đổi trong tư duy quản lý. Thuật ngữ “Document” trong ISO 13485 (Clause 0.2) giờ đây được hiểu là tập hợp của: Thiết lập (Define) + Thực hiện (Implement) + Duy trì (Maintain).
Ma trận đối chiếu (Mapping Matrix):
| Thuật ngữ cũ (QSR) | Thuật ngữ mới (QMSR/ISO 13485) | Điều khoản ISO 13485 | Ghi chú của Tư vấn |
| DHF (Design History File) | DDF (Design & Development File) | Clause 7.3.10 | Hồ sơ minh chứng quá trình thiết kế. |
| DMR (Device Master Record) | MDF (Medical Device File) | Clause 4.2.3 | “Blueprint” tổng thể cho sản phẩm. |
| DHR (Device History Record) | Batch Record / Device Record | Clause 7.5.1 | Bằng chứng sản xuất thực tế. |
| Management Representative | Top Management | Clause 5.1 | Chuyển trách nhiệm cho Ban lãnh đạo cao nhất. |
Lời khuyên thực thi: Doanh nghiệp có thể giữ lại các tên gọi cũ (DHF, DMR, DHR) trong hệ thống nội bộ để tránh xáo trộn lớn, nhưng bắt buộc phải lập một Ma trận đối chiếu (Mapping Matrix) trong Sổ tay Chất lượng để giải trình rõ ràng cho thanh tra FDA về sự tương đương giữa các thuật ngữ này với cấu trúc QMSR.
3. Hồ sơ Thiết kế và Phát triển (Design and Development File – DDF)
DDF (Clause 7.3.10) yêu cầu lưu trữ toàn bộ hồ sơ chứng minh thiết bị được phát triển đúng theo kế hoạch đã phê duyệt.
- Lợi ích của “Gia đình thiết bị” (Device Family): Khác với DHF cũ thường tập trung vào từng mã sản phẩm đơn lẻ, DDF cho phép quản lý theo “gia đình thiết bị”. Điều này giúp giảm bớt gánh nặng hành chính bằng cách nhóm các biến thể có cùng đặc điểm thiết kế cơ bản vào một bộ hồ sơ chung.
- Tích hợp Quản lý rủi ro: Đây là yêu cầu cốt lõi. Các đầu ra của quản lý rủi ro (ISO 14971) phải là đầu vào bắt buộc của thiết kế (Design Inputs). Mọi thay đổi thiết kế phải được đánh giá rủi ro lại trước khi thực hiện.
- Thành phần bắt buộc: Kế hoạch, đầu vào, đầu ra, xem xét (Review), kiểm tra xác nhận (Verification), xác nhận giá trị sử dụng (Validation) và hồ sơ chuyển giao thiết kế.
4. Tệp Thiết bị Y tế (Medical Device File – MDF)
MDF (Clause 4.2.3) là tài liệu chuẩn để sản xuất hàng loạt. So với DMR, MDF rộng hơn vì yêu cầu tham chiếu đến cả dữ liệu lâm sàng và hồ sơ quản lý rủi ro.
- Kiểm soát Nhãn mác và Bao bì (§820.45): FDA bổ sung yêu cầu khắt khe hơn ISO. Doanh nghiệp phải thực hiện và lưu trữ bằng chứng kiểm tra độ chính xác của nhãn mác (về thông tin, hạn dùng, mã UDI) trước khi xuất xưởng.
- Nội dung cốt lõi: Mô tả thiết bị, mục đích sử dụng, quy trình sản xuất, lắp đặt, bảo trì và các yêu cầu về đo lường, giám sát.
5. Hồ sơ Lịch sử Thiết bị và Các bản ghi bổ sung (§820.35)
Hồ sơ lô (Batch Records) cung cấp bằng chứng rằng sản phẩm được sản xuất đúng theo MDF. Ngoài các dữ liệu về ngày sản xuất, số lượng và UDI theo ISO, FDA yêu cầu các trường thông tin cụ thể sau:
A. Hồ sơ Khiếu nại (Complaints) – 7 trường bắt buộc:
- Tên thiết bị.
- Ngày nhận khiếu nại.
- Mã định danh thiết bị (UDI/UPC) và các nhận dạng khác.
- Tên, địa chỉ và số điện thoại của người khiếu nại.
- Bản chất và chi tiết của khiếu nại.
- Bất kỳ hành động khắc phục hoặc phòng ngừa nào đã thực hiện.
- Nội dung phản hồi cho người khiếu nại.
B. Hồ sơ Dịch vụ (Servicing) – 6 trường bắt buộc:
- Tên thiết bị được bảo trì.
- Mã UDI/UPC hoặc nhận dạng thiết bị.
- Ngày thực hiện dịch vụ.
- Tên (các) cá nhân thực hiện dịch vụ.
- Nội dung dịch vụ đã thực hiện.
- Dữ liệu kiểm tra và xác nhận sau dịch vụ.
Quyền bảo mật (§820.35(d)): Doanh nghiệp có quyền đánh dấu các hồ sơ này là “Confidential” (Bảo mật). Việc này giúp FDA áp dụng đúng các quy định về thông tin công cộng (Part 20) để bảo vệ bí mật thương mại của doanh nghiệp khi có yêu cầu truy cập dữ liệu từ bên ngoài.
6. Kiểm soát Tài liệu và Sự sụp đổ của “Bức tường lửa” §820.180(c)
Đây là thay đổi rủi ro nhất đối với doanh nghiệp.
- Mất ngoại lệ tiếp cận: FDA đã bãi bỏ §820.180(c). Hiện nay, các bản ghi đánh giá nội bộ, xem xét của lãnh đạo và đánh giá nhà cung cấp đều nằm trong phạm vi kiểm tra của FDA. Sẽ không còn “bức tường lửa” bảo vệ các báo cáo này. Nếu hồ sơ đánh giá nội bộ của bạn phát hiện lỗi mà không có CAPA tương ứng, đó sẽ là bằng chứng trực tiếp cho một 483 hoặc Warning Letter.
- Liêm chính dữ liệu (Data Integrity): Áp dụng 21 CFR Part 11 là bắt buộc đối với eQMS.
- Điểm mới về Phê duyệt (89 FR 7496): FDA đã loại bỏ yêu cầu bắt buộc phải có chữ ký và ngày tháng trên từng bản ghi phê duyệt nếu hệ thống điện tử của doanh nghiệp có Audit Trail (Nhật ký hệ thống) phù hợp để chứng minh tính xác thực. Đây là cơ hội lớn để tối ưu hóa quy trình số hóa.
7. Lộ trình Triển khai và Sẵn sàng Thanh tra (Inspection Readiness)
FDA sẽ ngừng sử dụng kỹ thuật QSIT (vốn tập trung vào 4 hệ thống chính) và chuyển sang Compliance Program 7382.850. Quy trình mới này sẽ đánh giá theo cấu trúc từng điều khoản của ISO 13485:2016 kết hợp với các yêu cầu bổ sung của FDA.
Khuyến nghị của Chuyên gia tư vấn:
- Phân tích khoảng cách (Gap Analysis): Rà soát không chỉ tài liệu mà còn cả năng lực của nhân sự trong việc vận hành quy trình mới.
- Cảnh giác với chứng chỉ ISO: Việc có chứng chỉ ISO 13485 từ tổ chức chứng nhận không thay thế cho thanh tra FDA. FDA không cấp chứng nhận; họ thực thi luật pháp.
- Thanh tra giả định (Mock Audit): Thực hiện các đợt đánh giá mô phỏng theo phương pháp mới của FDA, tập trung vào việc quản lý rủi ro và các bản ghi bổ sung tại §820.35.
Kết luận: Chuyển đổi sang QMSR không phải là một bài tập về thay đổi tên gọi tài liệu. Đó là sự nâng cấp toàn diện lên hệ thống quản lý rủi ro thực thụ. Doanh nghiệp cần hành động ngay để đảm bảo vị thế tại thị trường Hoa Kỳ, tránh rủi ro sản phẩm bị đình chỉ do vi phạm các yêu cầu về hồ sơ chất lượng.
Liên hệ để tư vấn doanh nghiệp
STC VN Co., Ltd. (Staunchly Vietnam)
Hotline: +84 933 096 426 – +84 868 591 260
Email: info@staunchlyservices.com.vn
Website: staunchlyservices.com.vn
Liên hệ tải tiêu chuẩn pdf song ngữ Anh Việt: chỉ từ 50$
Chúng tôi cung cấp tài liệu tiêu chuẩn bản gốc tiếng Anh và bản dịch tiếng Việt
• Bản gốc tiếng anh của Tiêu Chuẩn kèm sơ đồ tóm tắt và hình ảnh minh họa rõ ràng. Để quý khách thuận tiện đối chiếu theo từng điều khoản, dễ tra cứu và lập hồ sơ.
• Bản dịch tiếng Việt chuẩn chuyên ngành, dùng được ngay cho công việc.
• Ghi chú diễn giải và hướng dẫn áp dụng từ chuyên gia thực chiến.
• Giao nhanh dưới định dạng PDF/Word thuận tiện chỉnh sửa, lưu trữ.
Phạm vi tiêu chuẩn
Hệ thống quản lý ISO (9001, 14001, 45001, 37001…..); chứng nhận bền vững (ISCC, FSC, VFCS/PEFC….); dệt may và vật liệu tái chế (GRS, OCS, OEKO-TEX….); trách nhiệm xã hội và ESG (SMETA, EcoVadis, Fairtrade….) cùng nhiều tiêu chuẩn ngành khác theo yêu cầu.
Vì sao chọn chúng tôi
Tài liệu được biên dịch bởi đội ngũ trực tiếp tư vấn và đánh giá chứng nhận, nên bản dịch không chỉ đúng ngôn ngữ mà còn đúng tinh thần áp dụng thực tế — giúp doanh nghiệp tiết kiệm thời gian và hạn chế sai sót khi xây dựng hồ sơ.
Chi phí
Chỉ từ 50 USD/tài liệu, tùy độ dài và độ phức tạp. Quý Doanh nghiệp vui lòng gửi tên tiêu chuẩn để nhận báo giá chính xác miễn phí.
Cách đăng ký
1. Liên hệ và cho biết tên tiêu chuẩn cần dịch.
2. Nhận báo giá và thời gian giao tài liệu.
3. Thanh toán và nhận tài liệu qua email.
Thanh toán
Chuyển khoản qua mã QR ngân hàng (gửi kèm khi báo giá), hoặc qua PayPal: ducluongservices@gmail.com.
Rất mong được đồng hành cùng Quý Doanh nghiệp trên hành trình chứng nhận.
Trân trọng,
THÔNG TIN LIÊN HỆ
Hotline: +84 933 096 426 – +84 868 591 260